292:
the domain composition of interacting and non-interacting protein pairs. When given a protein pair to classify, RFD first creates a representation of the protein pair in a vector. The vector contains all the domain types used to train RFD, and for each domain type the vector also contains a value of 0, 1, or 2. If the protein pair does not contain a certain domain, then the value for that domain is 0. If one of the proteins of the pair contains the domain, then the value is 1. If both proteins contain the domain, then the value is 2. Using training data, RFD constructs a decision forest, consisting of many decision trees. Each decision tree evaluates several domains, and based on the presence or absence of interactions in these domains, makes a decision as to if the protein pair interacts. The vector representation of the protein pair is evaluated by each tree to determine if they are an interacting pair or a non-interacting pair. The forest tallies up all the input from the trees to come up with a final decision. The strength of this method is that it does not assume that domains interact independent of each other. This makes it so that multiple domains in proteins can be used in the prediction. This is a big step up from previous methods which could only predict based on a single domain pair. The limitation of this method is that it relies on the training dataset to produce results. Thus, usage of different training datasets could influence the results. A caveat of most methods is the lacks negative data, e.g non-interactions for proteins which can be overcome using topology-driven negative sampling.
148:) to build distance matrices for each of the proteins of interest. The distance matrices should then be used to build phylogenetic trees. However, comparisons between phylogenetic trees are difficult, and current methods circumvent this by simply comparing distance matrices. The distance matrices of the proteins are used to calculate a correlation coefficient, in which a larger value corresponds to co-evolution. The benefit of comparing distance matrices instead of phylogenetic trees is that the results do not depend on the method of tree building that was used. The downside is that difference matrices are not perfect representations of phylogenetic trees, and inaccuracies may result from using such a shortcut. Another factor worthy of note is that there are background similarities between the phylogenetic trees of any protein, even ones that do not interact. If left unaccounted for, this could lead to a high false-positive rate. For this reason, certain methods construct a background tree using 16S rRNA sequences which they use as the canonical tree of life. The distance matrix constructed from this tree of life is then subtracted from the distance matrices of the proteins of interest. However, because RNA distance matrices and DNA distance matrices have different scale, presumably because RNA and DNA have different mutation rates, the RNA matrix needs to be rescaled before it can be subtracted from the DNA matrices. By using molecular clock proteins, the scaling coefficient for protein distance/RNA distance can be calculated. This coefficient is used to rescale the RNA matrix.
152:
356:
calculates an E-score which measures if two domains interact. It is calculated as log(probability that the two proteins interact given that the domains interact/probability that the two proteins interact given that the domains don’t interact). The probabilities required in the formula are calculated using an
Expectation Maximization procedure, which is a method for estimating parameters in statistical models. High E-scores indicate that the two domains are likely to interact, while low scores indicate that other domains form the protein pair are more likely to be responsible for the interaction. The drawback with this method is that it does not take into account false positives and false negatives in the experimental data.
227:
314:
sequence-signature. They look specifically for sequence-signatures that are found together more often than by chance. This uses a log-odds score which is computed as log2(Pij/PiPj), where Pij is the observed frequency of domains i and j occurring in one protein pair; Pi and Pj are the background frequencies of domains i and j in the data. Predicted domain interactions are those with positive log-odds scores and also having several occurrences within the database. The downside with this method is that it looks at each pair of interacting domains separately, and it assumes that they interact independently of each other.
305:) to search for protein complex structures that are homologous to the query sequences. These known complex structures are then used as templates to structurally model the interaction between query sequences. This method has the advantage of not only inferring protein interactions but also suggests models of how proteins interact structurally, which can provide some insights into the atomic level mechanism of that interaction. On the other hand, the ability for these methods to make a prediction is constrained by a limited number of known protein complex structures.
377:, which attempts to use geometric and steric considerations to fit two proteins of known structure into a bound complex. This is a useful mode of inquiry in cases where both proteins in the pair have known structures and are known (or at least strongly suspected) to interact, but since so many proteins do not have experimentally determined structures, sequence-based interaction prediction methods are especially useful in conjunction with experimental studies of an organism's
105:
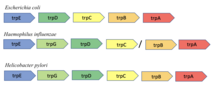
245:. Only the trpA and trpB genes are adjacent across all three organisms and are thus predicted to interact by the conserved gene neighborhood method. This image was adapted from Dandekar, T., Snel, B., Huynen, M., & Bork, P. (1998). Conservation of gene order: a fingerprint of proteins that physically interact.
211:
the beta similar to the second half. One limit of this method is that not all proteins that interact can be found fused in another genome, and therefore cannot be identified by this method. On the other hand, the fusion of two proteins does not necessitate that they physically interact. For instance, the
346:
integrate data from a wide variety of sources, including both experimental results and prior computational predictions, and use these features to assess the likelihood that a particular potential protein interaction is a true positive result. These methods are useful because experimental procedures,
262:
The conserved neighborhood method is based on the hypothesis that if genes encoding two proteins are neighbors on a chromosome in many genomes, then they are likely functionally related. The method is based on an observation by Bork et al. of gene pair conservation across nine bacterial and archaeal
210:
depicts the BLAST sequence alignment of
Succinyl coA Transferase with its two separate homologs in E. coli. The two subunits have non-overlapping regions of sequence similarity with the human protein, indicated by the pink regions, with the alpha subunit similar to the first half of the protein and
179:
is based on the hypothesis that interacting proteins are sometimes fused into a single protein. For instance, two or more separate proteins in a genome may be identified as fused into one single protein in another genome. The separate proteins are likely to interact and thus are likely functionally
158:
The Human succinyl-CoA-Transferase enzyme is represented by the two joint blue and green bars at the top of the image. The alpha subunit of the
Acetate-CoA-Transferase enzyme is homologous with the first half of the enzyme, represents by the blue bar. The beta subunit of the Acetate-CoA-Transferase
95:
Proteins that interact are more likely to co-evolve, therefore, it is possible to make inferences about interactions between pairs of proteins based on their phylogenetic distances. It has also been observed in some cases that pairs of interacting proteins have fused orthologues in other organisms.
291:
Classification methods use data to train a program (classifier) to distinguish positive examples of interacting protein/domain pairs with negative examples of non-interacting pairs. Popular classifiers used are Random Forest
Decision (RFD) and Support Vector Machines. RFD produces results based on
364:
The problem of PPI prediction can be framed as a supervised learning problem. In this paradigm the known protein interactions supervise the estimation of a function that can predict whether an interaction exists or not between two proteins given data about the proteins (e.g., expression levels of
355:
The domain-pair exclusion analysis detects specific domain interactions that are hard to detect using
Bayesian methods. Bayesian methods are good at detecting nonspecific promiscuous interactions and not very good at detecting rare specific interactions. The domain-pair exclusion analysis method
313:
Association methods look for characteristic sequences or motifs that can help distinguish between interacting and non-interacting pairs. A classifier is trained by looking for sequence-signature pairs where one protein contains one sequence-signature, and its interacting partner contains another
139:
It was observed that the phylogenetic trees of ligands and receptors were often more similar than due to random chance. This is likely because they faced similar selection pressures and co-evolved. This method uses the phylogenetic trees of protein pairs to determine if interactions exist. To do
330:
of the atoms involved. The sequences in the library are then clustered based on structural alignment and redundant sequences are eliminated. The residues that have a high (generally >50%) level of frequency for a given position are considered hotspots. This library is then used to identify
205:
is necessary. For example, if we had the amino acid sequences of proteins A and B and the amino acid sequences of all proteins in a certain genome, we could check each protein in that genome for non-overlapping regions of sequence similarity to both proteins A and B.
126:
illustrates a hypothetical situation in which proteins A and B are identified as functionally linked due to their identical phylogenetic profiles across 5 different genomes. The Joint Genome
Institute provides an Integrated Microbial Genomes and Microbiomes database
347:
particularly the yeast two-hybrid experiments, are extremely noisy and produce many false positives, while the previously mentioned computational methods can only provide circumstantial evidence that a particular pair of proteins might interact.
300:
This group of methods makes use of known protein complex structures to predict and structurally model interactions between query protein sequences. The prediction process generally starts by employing a sequence based method (e.g.
151:
111:
The phylogenetic profiles of four genes (A, B, C and D) are shown on the right. A '1' denotes presence of the gene in the genome and a '0' denotes absence. The two identical profiles of genes A and B are highlighted in
1088:
Jansen, R; Yu, H; Greenbaum, D; Kluger, Y; Krogan, NJ; Chung, S; Emili, A; Snyder, M; Greenblatt, JF; Gerstein, M (2003). "A Bayesian networks approach for predicting protein–protein interactions from genomic data".
96:
In addition, a number of bound protein complexes have been structurally solved and can be used to identify the residues that mediate the interaction so that similar motifs can be located in other organisms.
1404:
45:
is important for the investigation of intracellular signaling pathways, modelling of protein complex structures and for gaining insights into various biochemical processes.
283:
enzyme known to interact to catalyze a single reaction. The adjacency of these two genes was shown to be conserved across nine different bacterial and archaeal genomes.
839:
Fukuhara, Naoshi, and
Takeshi Kawabata. (2008) "HOMCOS: a server to predict interacting protein pairs and interacting sites by homology modeling of complex structures"
490:
Marcotte E.M., Pellegrini M., Ng H.L., Rice D.W., Yeates T.O., Eisenberg D. (1999) "Detecting protein function and protein–protein interactions from genome sequences."
1053:
Keskin, O.; Ma, B.; Nussinov, R. (2004). "Hot regions in protein–protein interactions: The organization and contribution of structurally conserved hot spot residues".
769:
Chatterjee, Ayan; Ravandi, Babak; Philip, Naomi H.; Abdelmessih, Mario; Mowrey, William R.; Ricchiuto, Piero; Liang, Yupu; Ding, Wei; Mobarec, Juan C. (2024-04-29),
638:
Pazos, F; Ranea, JA; Juan, D; Sternberg, MJ (2005). "Assessing protein coevolution in the context of the tree of life assists in the prediction of the interactome".
263:
genomes. The method is most effective in prokaryotes with operons as the organization of genes in an operon is generally related to function. For instance, the
122:
is based on the hypothesis that if two or more proteins are concurrently present or absent across several genomes, then they are likely functionally related.
425:
1150:
Zhang, QC; Petrey, D; Deng, L; Qiang, L; Shi, Y; Thu, CA; Bisikirska, B; Lefebvre, C; Accili, D; Hunter, T; Maniatis, T; Califano, A; Honig, B (2012).
1296:
1544:
856:
Kittichotirat W, M Guerquin, RE Bumgarner, and R Samudrala (2009) "Protinfo PPC: a web server for atomic level prediction of protein complexes"
470:
Enright A.J.,Iliopoulos I.,Kyripides N.C. and
Ouzounis C.A. (1999) "Protein interaction maps for complete genomes based on gene fusion events."
159:
enzyme is homologous with the second half of the enzyme, represents by the green bar. This mage was adapted from Uetz, P. & Pohl, E. (2018)
1539:
226:
56:
410:
457:
Dandekar T., Snel B.,Huynen M. and Bork P. (1998) "Conservation of gene order: a fingerprint of proteins that physically interact."
1394:
1374:
395:
42:
1355:
876:"Inferred Biomolecular Interaction Server--a web server to analyze and predict protein interacting partners and binding sites"
874:
Shoemaker, BA; Zhang, D; Thangudu, RR; Tyagi, M; Fong, JH; Marchler-Bauer, A; Bryant, SH; Madej, T; Panchenko, AR (Jan 2010).
1209:"Deciphering protein–protein interactions. Part II. Computational methods to predict protein and domain interaction partners"
1384:
1289:
326:, where the interfaces are defined as pairs of polypeptide fragments that are below a threshold slightly larger than the
1432:
405:
1531:
1437:
1365:
400:
963:"Prediction of protein–protein interactions by combining structure and sequence conservation in protein interfaces"
1389:
1379:
1369:
1331:
331:
potential interactions between pairs of targets, providing that they have a known structure (i.e. present in the
68:
615:
Tan S.H., Zhang Z., Ng S.K. (2004) "ADVICE: Automated
Detection and Validation of Interaction by Co-Evolution."
51:, physical interactions between pairs of proteins can be inferred from a variety of techniques, including yeast
1409:
1282:
223:
are known to interact. However, many proteins possess homologs of these domains and they do not all interact.
41:
in an attempt to identify and catalog physical interactions between pairs or groups of proteins. Understanding
926:
Sprinzak, E; Margalit, H (2001). "Correlated sequence-signatures as markers of protein–protein interaction".
676:
Dandekar, T. (1998-09-01). "Conservation of gene order: a fingerprint of proteins that physically interact".
374:
186:
1480:
1442:
1106:
420:
117:
1399:
201:
141:
75:
of numerous species are ongoing. Experimentally determined interactions usually provide the basis for
17:
1447:
1326:
1220:
1163:
1098:
772:
Topology-Driven
Negative Sampling Enhances Generalizability in Protein-Protein Interaction Prediction
430:
327:
52:
1111:
1565:
1414:
1360:
1351:
365:
each gene in different experimental conditions, location information, phylogenetic profile, etc.).
279:
1346:
1132:
80:
64:
38:
199:. In order to identify these sequences, a sequence similarity algorithm such as the one used by
104:
1269:
1501:
1248:
1189:
1124:
1070:
1035:
984:
943:
905:
822:
770:
751:
701:
693:
655:
598:
580:
536:
332:
323:
220:
60:
1516:
1238:
1228:
1179:
1171:
1116:
1062:
1025:
1015:
974:
935:
895:
887:
812:
776:
741:
685:
647:
588:
570:
526:
273:
83:
protein sequences across species. However, there are also methods that predict interactions
140:
this, homologs of the proteins of interest are found (using a sequence search tool such as
1485:
1309:
415:
343:
1224:
1167:
1102:
1424:
1341:
1243:
1208:
1184:
1151:
1030:
1003:
900:
875:
593:
558:
373:
The field of protein–protein interaction prediction is closely related to the field of
34:
817:
800:
689:
1559:
1521:
1511:
1452:
979:
962:
746:
729:
1506:
1475:
1336:
1152:"Structure-based prediction of protein–protein interactions on a genome-wide scale"
1136:
730:"Prediction of protein–protein interactions using random decision forest framework"
163:. In: Wink, M. (ed.), Introduction to Molecular Biotechnology, 3rd ed. Wiley-VCH,
1274:
1233:
435:
390:
378:
72:
531:
514:
131:) that has a phylogenetic profiling tool for single genes and gene cassettes.
1318:
1066:
780:
651:
515:"Similarity of phylogenetic trees as indicator of protein–protein interaction"
216:
212:
184:
enzyme, which is found as one protein in humans but as two separate proteins,
697:
584:
1120:
302:
1252:
1193:
1128:
1074:
1039:
988:
947:
939:
909:
826:
755:
659:
602:
540:
705:
575:
322:
This method builds a library of known protein–protein interfaces from the
144:) and multiple-sequence alignments are done (with alignment tools such as
135:
Prediction of co-evolved protein pairs based on similar phylogenetic trees
1020:
891:
1175:
1305:
801:"InterPreTS: protein Interaction Prediction through Tertiary Structure"
233:
Organization of the trp operon in three different species of bacteria:
145:
1002:
Ogmen, U.; Keskin, O.; Aytuna, A.S.; Nussinov, R.; Gursoy, A. (2005).
559:"Construction and analysis of protein–protein interaction networks"
1467:
225:
150:
103:
1278:
128:
67:, fluorescence resonance energy transfer (FRET), and
1004:"PRISM: protein interactions by structural matching"
296:
Inference of interactions from homologous structures
87:, without prior knowledge of existing interactions.
1530:
1494:
1466:
1423:
1317:
71:(MST). Efforts to experimentally determine the
961:Aytuna, A. S.; Keskin, O.; Gursoy, A. (2005).
1290:
8:
633:
631:
629:
161:Protein–Protein and Protein–DNA Interactions
921:
919:
1297:
1283:
1275:
486:
484:
482:
480:
453:
451:
1270:Overview of protein interaction databases
1242:
1232:
1183:
1110:
1029:
1019:
978:
899:
816:
745:
592:
574:
530:
177:The Rosetta Stone or Domain Fusion method
27:Prediction by observation and computation
508:
506:
504:
502:
500:
447:
57:protein-fragment complementation assays
1540:Photoactivated localization microscopy
1458:Protein–protein interaction prediction
723:
721:
719:
717:
715:
426:Protein–DNA interaction site predictor
31:Protein–protein interaction prediction
18:Protein-protein interaction prediction
1207:Shoemaker, BA; Panchenko, AR (2007).
411:Protein structure prediction software
318:Identification of structural patterns
7:
671:
669:
552:
550:
79:to predict interactions, e.g. using
1415:Freeze-fracture electron microscopy
180:related. An example of this is the
172:Rosetta stone (gene fusion) method
25:
799:Aloy, P.; Russell, R. B. (2003).
1395:Isothermal titration calorimetry
1375:Dual-polarization interferometry
513:Pazos, F.; Valencia, A. (2001).
818:10.1093/bioinformatics/19.1.161
369:Relationship to docking methods
277:encode the two subunits of the
1014:(Web Server issue): W331–336.
678:Trends in Biochemical Sciences
351:Domain-pair exclusion analysis
247:Trends in biochemical sciences
182:Human Succinyl coA Transferase
1:
1385:Chromatin immunoprecipitation
980:10.1093/bioinformatics/bti443
747:10.1093/bioinformatics/bti721
690:10.1016/S0968-0004(98)01274-2
557:Raman, Karthik (2010-02-15).
59:(PCA), affinity purification/
1448:Protein structural alignment
1433:Protein structure prediction
1234:10.1371/journal.pcbi.0030043
406:Protein structure prediction
193:Acetate coA Transferase beta
43:protein–protein interactions
1532:Super-resolution microscopy
1438:Protein function prediction
1366:Peptide mass fingerprinting
1361:Protein immunoprecipitation
886:(Database issue): D518–24.
864:(Web Server issue): 519-25.
401:Protein function prediction
396:Protein–protein interaction
360:Supervised learning problem
258:Conserved gene neighborhood
1582:
623:(Web Server issue):W69-72.
339:Bayesian network modelling
1390:Surface plasmon resonance
1380:Microscale thermophoresis
1370:Protein mass spectrometry
1332:Green fluorescent protein
1067:10.1016/j.jmb.2004.10.077
781:10.1101/2024.04.27.591478
728:Chen, XW; Liu, M (2005).
652:10.1016/j.jmb.2005.07.005
563:Automated Experimentation
69:Microscale Thermophoresis
1410:Cryo-electron microscopy
532:10.1093/protein/14.9.609
118:The phylogenetic profile
1443:Protein–protein docking
1356:Protein electrophoresis
1121:10.1126/science.1087361
375:protein–protein docking
187:Acetate coA Transferase
1342:Protein immunostaining
940:10.1006/jmbi.2001.4920
858:Nucleic Acids Research
841:Nucleic Acids Research
421:Macromolecular docking
287:Classification methods
254:
239:Haemophilus influenzae
168:
113:
100:Phylogenetic profiling
1400:X-ray crystallography
576:10.1186/1759-4499-2-2
229:
154:
107:
77:computational methods
33:is a field combining
1327:Protein purification
459:Trends Biochem. Sci.
431:Two-hybrid screening
328:Van der Waals radius
1352:Gel electrophoresis
1225:2007PLSCB...3...43S
1176:10.1038/nature11503
1168:2012Natur.490..556Z
1103:2003Sci...302..449J
519:Protein Engineering
309:Association methods
280:tryptophan synthase
243:Helicobacter pylori
65:protein microarrays
1495:Display techniques
1347:Protein sequencing
1021:10.1093/nar/gki585
892:10.1093/nar/gkp842
617:Nucleic Acids Res.
255:
169:
114:
39:structural biology
1553:
1552:
1502:Bacterial display
1008:Nucleic Acids Res
973:(12): 2850–2855.
880:Nucleic Acids Res
740:(24): 4394–4400.
61:mass spectrometry
16:(Redirected from
1573:
1517:Ribosome display
1453:Protein ontology
1299:
1292:
1285:
1276:
1257:
1256:
1246:
1236:
1213:PLOS Comput Biol
1204:
1198:
1197:
1187:
1162:(7421): 556–60.
1147:
1141:
1140:
1114:
1097:(5644): 449–53.
1085:
1079:
1078:
1061:(5): 1281–1294.
1050:
1044:
1043:
1033:
1023:
999:
993:
992:
982:
958:
952:
951:
923:
914:
913:
903:
871:
865:
854:
848:
837:
831:
830:
820:
796:
790:
789:
788:
787:
766:
760:
759:
749:
725:
710:
709:
673:
664:
663:
646:(4): 1002–1015.
635:
624:
613:
607:
606:
596:
578:
554:
545:
544:
534:
510:
495:
488:
475:
468:
462:
455:
344:Bayesian methods
274:Escherichia coli
235:Escherichia coli
197:Escherichia coli
21:
1581:
1580:
1576:
1575:
1574:
1572:
1571:
1570:
1556:
1555:
1554:
1549:
1526:
1490:
1486:Secretion assay
1462:
1419:
1313:
1303:
1266:
1261:
1260:
1206:
1205:
1201:
1149:
1148:
1144:
1112:10.1.1.217.8151
1087:
1086:
1082:
1052:
1051:
1047:
1001:
1000:
996:
960:
959:
955:
925:
924:
917:
873:
872:
868:
855:
851:
838:
834:
798:
797:
793:
785:
783:
768:
767:
763:
727:
726:
713:
675:
674:
667:
637:
636:
627:
614:
610:
556:
555:
548:
525:(14): 609–614.
512:
511:
498:
489:
478:
469:
465:
456:
449:
444:
416:Gene prediction
387:
371:
362:
353:
341:
320:
311:
298:
289:
260:
219:domains in the
174:
137:
102:
93:
28:
23:
22:
15:
12:
11:
5:
1579:
1577:
1569:
1568:
1558:
1557:
1551:
1550:
1548:
1547:
1542:
1536:
1534:
1528:
1527:
1525:
1524:
1519:
1514:
1509:
1504:
1498:
1496:
1492:
1491:
1489:
1488:
1483:
1478:
1472:
1470:
1464:
1463:
1461:
1460:
1455:
1450:
1445:
1440:
1435:
1429:
1427:
1425:Bioinformatics
1421:
1420:
1418:
1417:
1412:
1407:
1402:
1397:
1392:
1387:
1382:
1377:
1372:
1363:
1358:
1349:
1344:
1339:
1334:
1329:
1323:
1321:
1315:
1314:
1304:
1302:
1301:
1294:
1287:
1279:
1273:
1272:
1265:
1264:External links
1262:
1259:
1258:
1199:
1142:
1080:
1045:
994:
967:Bioinformatics
953:
934:(4): 681–692.
915:
866:
849:
832:
811:(1): 161–162.
805:Bioinformatics
791:
761:
734:Bioinformatics
711:
684:(9): 324–328.
665:
625:
608:
546:
496:
494:(285), 751-753
476:
463:
446:
445:
443:
440:
439:
438:
433:
428:
423:
418:
413:
408:
403:
398:
393:
386:
383:
370:
367:
361:
358:
352:
349:
340:
337:
319:
316:
310:
307:
297:
294:
288:
285:
259:
256:
173:
170:
136:
133:
101:
98:
92:
89:
49:Experimentally
35:bioinformatics
26:
24:
14:
13:
10:
9:
6:
4:
3:
2:
1578:
1567:
1564:
1563:
1561:
1546:
1543:
1541:
1538:
1537:
1535:
1533:
1529:
1523:
1522:Yeast display
1520:
1518:
1515:
1513:
1512:Phage display
1510:
1508:
1505:
1503:
1500:
1499:
1497:
1493:
1487:
1484:
1482:
1481:Protein assay
1479:
1477:
1474:
1473:
1471:
1469:
1465:
1459:
1456:
1454:
1451:
1449:
1446:
1444:
1441:
1439:
1436:
1434:
1431:
1430:
1428:
1426:
1422:
1416:
1413:
1411:
1408:
1406:
1403:
1401:
1398:
1396:
1393:
1391:
1388:
1386:
1383:
1381:
1378:
1376:
1373:
1371:
1367:
1364:
1362:
1359:
1357:
1353:
1350:
1348:
1345:
1343:
1340:
1338:
1335:
1333:
1330:
1328:
1325:
1324:
1322:
1320:
1316:
1311:
1307:
1300:
1295:
1293:
1288:
1286:
1281:
1280:
1277:
1271:
1268:
1267:
1263:
1254:
1250:
1245:
1240:
1235:
1230:
1226:
1222:
1218:
1214:
1210:
1203:
1200:
1195:
1191:
1186:
1181:
1177:
1173:
1169:
1165:
1161:
1157:
1153:
1146:
1143:
1138:
1134:
1130:
1126:
1122:
1118:
1113:
1108:
1104:
1100:
1096:
1092:
1084:
1081:
1076:
1072:
1068:
1064:
1060:
1056:
1049:
1046:
1041:
1037:
1032:
1027:
1022:
1017:
1013:
1009:
1005:
998:
995:
990:
986:
981:
976:
972:
968:
964:
957:
954:
949:
945:
941:
937:
933:
929:
922:
920:
916:
911:
907:
902:
897:
893:
889:
885:
881:
877:
870:
867:
863:
859:
853:
850:
846:
842:
836:
833:
828:
824:
819:
814:
810:
806:
802:
795:
792:
782:
778:
774:
773:
765:
762:
757:
753:
748:
743:
739:
735:
731:
724:
722:
720:
718:
716:
712:
707:
703:
699:
695:
691:
687:
683:
679:
672:
670:
666:
661:
657:
653:
649:
645:
641:
634:
632:
630:
626:
622:
618:
612:
609:
604:
600:
595:
590:
586:
582:
577:
572:
568:
564:
560:
553:
551:
547:
542:
538:
533:
528:
524:
520:
516:
509:
507:
505:
503:
501:
497:
493:
487:
485:
483:
481:
477:
473:
467:
464:
460:
454:
452:
448:
441:
437:
434:
432:
429:
427:
424:
422:
419:
417:
414:
412:
409:
407:
404:
402:
399:
397:
394:
392:
389:
388:
384:
382:
380:
376:
368:
366:
359:
357:
350:
348:
345:
338:
336:
334:
329:
325:
317:
315:
308:
306:
304:
295:
293:
286:
284:
282:
281:
276:
275:
270:
266:
257:
253:(9), 324-328.
252:
248:
244:
240:
236:
232:
228:
224:
222:
218:
214:
209:
204:
203:
198:
194:
190:
188:
183:
178:
171:
166:
162:
157:
153:
149:
147:
143:
134:
132:
130:
125:
121:
119:
110:
106:
99:
97:
90:
88:
86:
82:
78:
74:
70:
66:
62:
58:
54:
50:
46:
44:
40:
36:
32:
19:
1507:mRNA display
1476:Enzyme assay
1457:
1337:Western blot
1319:Experimental
1216:
1212:
1202:
1159:
1155:
1145:
1094:
1090:
1083:
1058:
1055:J. Mol. Biol
1054:
1048:
1011:
1007:
997:
970:
966:
956:
931:
927:
883:
879:
869:
861:
857:
852:
844:
840:
835:
808:
804:
794:
784:, retrieved
771:
764:
737:
733:
681:
677:
643:
639:
620:
616:
611:
566:
562:
522:
518:
491:
474:(402), 86-90
471:
466:
461:(23),324-328
458:
372:
363:
354:
342:
321:
312:
299:
290:
278:
272:
268:
264:
261:
250:
246:
242:
238:
234:
230:
207:
200:
196:
192:
185:
181:
176:
175:
164:
160:
155:
138:
123:
116:
115:
108:
94:
84:
76:
48:
47:
30:
29:
1545:Vertico SMI
1405:Protein NMR
847:(S2): 185-.
436:FastContact
391:Interactome
379:interactome
221:src protein
73:interactome
1566:Proteomics
1219:(4): e43.
928:J Mol Biol
786:2024-05-04
640:J Mol Biol
442:References
81:homologous
53:two-hybrid
1107:CiteSeerX
698:0968-0004
585:1759-4499
303:Interolog
271:genes in
156:Figure B.
109:Figure A.
55:systems,
1560:Category
1312:of study
1306:Proteins
1253:17465672
1194:23023127
1129:14564010
1075:15644221
1040:15991339
989:15855251
948:11518523
910:19843613
827:12499311
756:16234318
660:16139301
603:20334628
569:(1): 2.
541:11707606
385:See also
231:FigureC.
208:Figure B
165:in press
124:Figure A
1310:methods
1244:1857810
1221:Bibcode
1185:3482288
1164:Bibcode
1137:5293611
1099:Bibcode
1091:Science
1031:1160261
901:2808861
706:9787636
594:2834675
492:Science
146:Clustal
129:JGI IMG
112:yellow.
91:Methods
85:de novo
1308:: key
1251:
1241:
1192:
1182:
1156:Nature
1135:
1127:
1109:
1073:
1038:
1028:
987:
946:
908:
898:
825:
754:
704:
696:
658:
601:
591:
583:
539:
472:Nature
120:method
1468:Assay
1133:S2CID
202:BLAST
195:, in
189:alpha
142:BLAST
1249:PMID
1190:PMID
1125:PMID
1071:PMID
1036:PMID
985:PMID
944:PMID
906:PMID
823:PMID
752:PMID
702:PMID
694:ISSN
656:PMID
599:PMID
581:ISSN
537:PMID
269:trpB
267:and
265:trpA
215:and
191:and
37:and
1239:PMC
1229:doi
1180:PMC
1172:doi
1160:490
1117:doi
1095:302
1063:doi
1059:345
1026:PMC
1016:doi
975:doi
936:doi
932:311
896:PMC
888:doi
813:doi
777:doi
742:doi
686:doi
648:doi
644:352
589:PMC
571:doi
527:doi
335:).
333:PDB
324:PDB
217:SH3
213:SH2
1562::
1247:.
1237:.
1227:.
1215:.
1211:.
1188:.
1178:.
1170:.
1158:.
1154:.
1131:.
1123:.
1115:.
1105:.
1093:.
1069:.
1057:.
1034:.
1024:.
1012:33
1010:.
1006:.
983:.
971:21
969:.
965:.
942:.
930:.
918:^
904:.
894:.
884:38
882:.
878:.
862:37
860:,
845:36
843:,
821:.
809:19
807:.
803:.
775:,
750:.
738:21
736:.
732:.
714:^
700:.
692:.
682:23
680:.
668:^
654:.
642:.
628:^
621:32
619:,
597:.
587:.
579:.
565:.
561:.
549:^
535:.
521:.
517:.
499:^
479:^
450:^
381:.
251:23
249:,
241:,
237:,
63:,
1368:/
1354:/
1298:e
1291:t
1284:v
1255:.
1231::
1223::
1217:3
1196:.
1174::
1166::
1139:.
1119::
1101::
1077:.
1065::
1042:.
1018::
991:.
977::
950:.
938::
912:.
890::
829:.
815::
779::
758:.
744::
708:.
688::
662:.
650::
605:.
573::
567:2
543:.
529::
523:9
167:.
127:(
20:)
Text is available under the Creative Commons Attribution-ShareAlike License. Additional terms may apply.