475:. This new sequencing method generated reads much shorter than those of Sanger sequencing: initially about 100 bases, now 400-500 bases. Its much higher throughput and lower cost (compared to Sanger sequencing) pushed the adoption of this technology by genome centers, which in turn pushed development of sequence assemblers that could efficiently handle the read sets. The sheer amount of data coupled with technology-specific error patterns in the reads delayed development of assemblers; at the beginning in 2004 only the
520:) continue to emerge. Despite the higher error rates of these technologies they are important for assembly because their longer read length helps to address the repeat problem. It is impossible to assemble through a perfect repeat that is longer than the maximum read length; however, as reads become longer the chance of a perfect repeat that large becomes small. This gives longer sequencing reads an advantage in assembling repeats even if they have low accuracy (~85%).
178:
33:
493:(previously Solexa) technology has been available and can generate about 100 million reads per run on a single sequencing machine. Compare this to the 35 million reads of the human genome project which needed several years to be produced on hundreds of sequencing machines. Illumina was initially limited to a length of only 36 bases, making it less suitable for de novo assembly (such as
442:) was invented and until shortly after 2000, the technology was improved up to a point where fully automated machines could churn out sequences in a highly parallelised mode 24 hours a day. Large genome centers around the world housed complete farms of these sequencing machines, which in turn led to the necessity of assemblers to be optimised for sequences from whole-genome
497:), but newer iterations of the technology achieve read lengths above 100 bases from both ends of a 3-400bp clone. Announced at the end of 2007, the SHARCGS assembler by Dohm et al. was the first published assembler that was used for an assembly with Solexa reads. It was quickly followed by a number of others.
304:) to 3 billion (e.g., the human genome) base pairs. Subsequent to these efforts, several other groups, mostly at the major genome sequencing centers, built large-scale assemblers, and an open source effort known as AMOS was launched to bring together all the innovations in genome assembly technology under the
710:. For example, sequencing "NAAAAAAAAAAAAN" and "NAAAAAAAAAAAN" which include 12 adenine might be wrongfully called with 11 adenine instead. Sequencing a highly repetitive segment of the target DNA/RNA might result in a call that is one base shorter or one base longer. Read quality is typically measured by
479:
assembler from 454 was available. Released in mid-2007, the hybrid version of the MIRA assembler by
Chevreux et al. was the first freely available assembler that could assemble 454 reads as well as mixtures of 454 reads and Sanger reads. Assembling sequences from different sequencing technologies was
422:
The complexity of sequence assembly is driven by two major factors: the number of fragments and their lengths. While more and longer fragments allow better identification of sequence overlaps, they also pose problems as the underlying algorithms show quadratic or even exponential complexity behaviour
730:
Assembly: During this step, reads alignment will be utilized with different criteria to map each read to the possible location. The predicted position of a read is based on either how much of its sequence aligns with other reads or a reference. Different alignment algorithms are used for reads from
315:
Strategy how a sequence assembler would take fragments (shown below the black bar) and match overlaps among them to assembly the final sequence (in black). Potentially problematic repeats are shown above the sequence (in pink above). Without overlapping fragments it may be impossible to assign these
224:
advantages (i.e. call quality). The logic behind it is to group the reads by smaller windows within the reference. Reads in each group will then be reduced in size using the k-mere approach to select the highest quality and most probable contiguous (contig). Contigs will then will be joined together
168:
The problem of sequence assembly can be compared to taking many copies of a book, passing each of them through a shredder with a different cutter, and piecing the text of the book back together just by looking at the shredded pieces. Besides the obvious difficulty of this task, there are some extra
402:
Referring to the comparison drawn to shredded books in the introduction: while for mapping assemblies one would have a very similar book as a template (perhaps with the names of the main characters and a few locations changed), de-novo assemblies present a more daunting challenge in that one would
333:
of a cell and represent only a subset of the whole genome. A number of algorithmical problems differ between genome and EST assembly. For instance, genomes often have large amounts of repetitive sequences, concentrated in the intergenic regions. Transcribed genes contain many fewer repeats, making
373:
In terms of complexity and time requirements, de-novo assemblies are orders of magnitude slower and more memory intensive than mapping assemblies. This is mostly due to the fact that the assembly algorithm needs to compare every read with every other read (an operation that has a naive time
328:
or EST assembly was an early strategy, dating from the mid-1990s to the mid-2000s, to assemble individual genes rather than whole genomes. The problem differs from genome assembly in several ways. The input sequences for EST assembly are fragments of the transcribed
463:
With the Sanger technology, bacterial projects with 20,000 to 200,000 reads could easily be assembled on one computer. Larger projects, like the human genome with approximately 35 million reads, needed large computing farms and distributed computing.
397:
approach, which may also use one of the OLC or DBG approaches. With greedy graph-based algorithms, the contigs, series of reads aligned together, grow by greedy extension, always taking on the read that is found by following the highest-scoring
312:
423:
to both number of fragments and their length. And while shorter sequences are faster to align, they also complicate the layout phase of an assembly as shorter reads are more difficult to use with repeats or near identical repeats.
203:
Reference-guided: grouping of reads by similarity to the most similar region within the reference (step wise mapping). Reads within each group are then shortened down to mimic short reads quality. A typical method to do so is the
169:
practical issues: the original may have many repeated paragraphs, and some shreds may be modified during shredding to have typos. Excerpts from another book may also be added in, and some shreds may be completely unrecognizable.
426:
In the earliest days of DNA sequencing, scientists could only gain a few sequences of short length (some dozen bases) after weeks of work in laboratories. Hence, these sequences could be aligned in a few minutes by hand.
149:
technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from
651:
Different organisms have a distinct region of higher complexity within their genome. Hence, the need of different computational approaches is needed. Some of the commonly used algorithms are:
703:
Pre-assembly: This step is essential to ensure the integrity of downstream analysis such as variant calling or final scaffold sequence. This step consists of two chronological workflows:
391:(DBG) approach, which is most widely applied to the short reads from the Solexa and SOLiD platforms. It relies on K-mer graphs, which performs well with vast quantities of short reads;
1260:
Li Z, Chen Y, Mu D, Yuan J, Shi Y, Zhang H, et al. (January 2012). "Comparison of the two major classes of assembly algorithms: overlap-layout-consensus and de-bruijn-graph".
300:
in 2000 and the human genome just a year later,—scientists developed assemblers like Celera
Assembler and Arachne able to handle genomes of 130 million (e.g., the fruit fly
1503:
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (October 2015). "BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs".
403:
not know beforehand whether this would become a science book, a novel, a catalogue, or even several books. Also, every shred would be compared with every other shred.
743:. On the other hand, algorithms aligning 3rd generation sequencing reads requires advance approaches to account for the high error rate associated with them.
532:. However, such measures do not assess assembly completeness in terms of gene content. Some tools evaluate the quality of an assembly after the fact.
200:
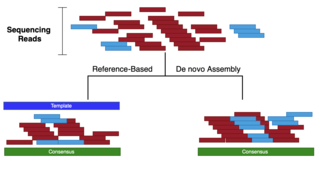
Mapping/Aligning: assembling reads by aligning reads against a template (AKA reference). The assembled consensus may not be identical to the template.
666:
Greedy Graph
Assembly: this approach score each added read to the assembly and selects the highest possible score from the overlapping region.
414:. On the other hand, in a mapping assembly, parts with multiple or no matches are usually left for another assembling technique to look into.
539:, using the fact that many genes are present only as single-copy genes in most genomes. The initial BUSCO sets represented 3023 genes for
768:
50:
1088:
Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, et al. (March 2000). "A whole-genome assembly of
Drosophila".
354:
116:
948:
97:
1662:
916:
This is useful post-assembly. It can generate different statistics and perform multiple filtering steps to the alignment file.
535:
For instance, BUSCO (Benchmarking
Universal Single-Copy Orthologs) is a measure of gene completeness in a genome, gene set, or
410:
representing neighboring repeats. Such information can be derived from reading a long fragment covering the repeats in full or
69:
938:
670:
Given a set of sequence fragments, the object is to find a longer sequence that contains all the fragments (see figure under
494:
437:
362:
350:
194:
54:
76:
861:
739:, quality, and the sequencing technique used plays a major role in choosing the best alignment algorithm in the case of
706:
Quality check: Depending on the type of sequencing technology, different errors might arise that would lead to a false
1225:
Nagaraj SH, Gasser RB, Ranganathan S (January 2007). "A hitchhiker's guide to expressed sequence tag (EST) analysis".
928:
764:
221:
209:
190:
189:
De-novo: assembling sequencing reads to create full-length (sometimes novel) sequences, without using a template (see
714:
which is an encoded score of each nucleotide quality within a read's sequence. Some sequencing technologies such as
338:), which means that unlike whole-genome shotgun sequencing, the reads are not uniformly sampled across the genome.
83:
43:
880:
This tool is designed to assemble (reference-guided) viral genomes at a greater accuracy using PacBio CCS reads.
334:
assembly somewhat easier. On the other hand, some genes are expressed (transcribed) in very high numbers (e.g.,
431:
158:
65:
242:
programs to piece together vast quantities of fragments generated by automated sequencing instruments called
1657:
619:
296:
1367:
Hu T, Chitnis N, Monos D, Dinh A (November 2021). "Next-generation sequencing technologies: An overview".
1105:
837:
481:
325:
162:
1453:"The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants"
953:
238:
The first sequence assemblers began to appear in the late 1980s and early 1990s as variants of simpler
378:)). Current de-novo genome assemblers may use different types of graph-based algorithms, such as the:
1097:
747:
746:
Post-assembly: This step is focusing on extracting valuable information from the assembled sequence.
501:
342:
305:
217:
1110:
817:
736:
711:
517:
284:) which can, in the worst case, increase the time and space complexity of algorithms quadratically;
1404:"SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing"
718:
do not have a scoring method for their sequenced reads. A common tool used in this step is FastQC.
1298:
Harrington CT, Lin EI, Olson MT, Eshleman JR (September 2013). "Fundamentals of pyrosequencing".
1131:
933:
808:
This is a common tool used to check reads quality from different sequencing technologies such as
732:
505:
490:
443:
411:
361:
was invented, EST sequencing was replaced by this far more efficient technology, described under
239:
151:
138:
385:(OLC) approach, which was typical of the Sanger-data assemblers and relies on an overlap graph;
294:
Faced with the challenge of assembling the first larger eukaryotic genomes—the fruit fly
177:
90:
1634:
1569:
1520:
1482:
1433:
1384:
1315:
1277:
1242:
1180:
1123:
1036:
994:
943:
885:
813:
472:
335:
274:
1587:
1149:
Batzoglou S, Jaffe DB, Stanley K, Butler J, Gnerre S, Mauceli E, et al. (January 2002).
803:
774:
Some of the common tools used in different assembly steps are listed in the following table:
1624:
1559:
1551:
1512:
1472:
1464:
1423:
1415:
1376:
1336:
1307:
1269:
1234:
1170:
1162:
1115:
1028:
986:
875:
731:
different sequencing technologies. Some of the commonly used approaches in the assembly are
529:
1588:"Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data"
977:
Sohn JI, Nam JW (January 2018). "The present and future of de novo whole-genome assembly".
721:
Filtering of reads: Reads that failed to pass the quality check should be removed from the
656:
513:
454:
287:
1101:
225:
to create a scaffold. The final consense is made by closing any gaps in the scaffold.
216:
Referenced-guided assembly is a combination of the other types. This type is applied on
1564:
1539:
1477:
1452:
1428:
1403:
809:
740:
468:
346:
263:
243:
146:
130:
1175:
1150:
1651:
1060:
536:
330:
1629:
1612:
1516:
528:
Most sequence assemblers have some algorithms built in for quality control, such as
1371:. Next Generation Sequencing and its Application to Medical Laboratory Immunology.
1135:
722:
707:
407:
1613:"Comparative analysis of algorithms for next-generation sequencing read alignment"
1119:
831:
699:
In general, there are three steps in assembling sequencing reads into a scaffold:
1380:
509:
32:
17:
1311:
1198:
540:
290:
in the fragments from the sequencing instruments, which can confound assembly.
259:
1040:
1016:
556:
544:
270:
1638:
1573:
1524:
1486:
1437:
1388:
1319:
1281:
1246:
1184:
1127:
998:
1468:
1273:
145:
sequence in order to reconstruct the original sequence. This is needed as
1238:
990:
903:
255:
251:
840:
tool. Mostly known for lightweight run and accurate sequence alignment.
311:
1419:
1032:
476:
358:
247:
1166:
893:
1555:
857:
715:
660:
552:
548:
246:. As the sequenced organisms grew in size and complexity (from small
154:
406:
Handling repeats in de-novo assembly requires the construction of a
769:
List of sequence alignment software § Short-read sequence alignment
341:
EST assembly is made much more complicated by features like (cis-)
851:
655:
Graph
Assembly: is based on Graph theory in computer science. The
592:
310:
205:
750:
and population analysis are examples of post-assembly analysis.
559:. This table shows an example for human and fruit fly genomes:
1402:
Dohm JC, Lottaz C, Borodina T, Himmelbauer H (November 2007).
1061:"De novo genome assembly versus mapping to a reference genome"
142:
26:
911:
1451:
Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM (April 2010).
691:
The result might not be an optimal solution to the problem.
1017:"De novo genome assembly: what every biologist should know"
804:
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
185:
There are three approaches to assembling sequencing data:
876:
https://github.com/salvocamiolo/LoReTTA/releases/tag/v0.1
208:
approach. Reference-guided assembly is most useful using
898:
This is an assembly tool that runs on the command line.
266:
needed increasingly sophisticated strategies to handle:
563:
BUSCO notation assessment results (C,D,F, M in %)
1611:
Ruffalo M, LaFramboise T, Koyutürk M (October 2011).
1337:"MIRA 2.9.8 for 454 and 454 / Sanger hybrid assembly"
1331:
1329:
687:
Repeat step 2 and 3 until only one fragment is left.
280:identical and nearly identical sequences (known as
57:. Unsourced material may be challenged and removed.
1540:"How to apply de Bruijn graphs to genome assembly"
1538:Compeau PE, Pevzner PA, Tesler G (November 2011).
1068:University of Applied Sciences Western Switzerland
1293:
1291:
453:contain sequencing artifacts like sequencing and
1300:Archives of Pathology & Laboratory Medicine
832:https://sourceforge.net/projects/bio-bwa/files/
678:Сalculate pairwise alignments of all fragments.
1220:
1218:
681:Choose two fragments with the largest overlap.
1498:
1496:
856:This command line tool is designed to handle
8:
1010:
1008:
659:is an example of this approach and utilizes
471:had been brought to commercial viability by
273:of sequencing data which need processing on
1151:"ARACHNE: a whole-genome shotgun assembler"
972:
970:
968:
1362:
1360:
1358:
1356:
1628:
1563:
1476:
1427:
1174:
1109:
516:were released and new technologies (e.g.
117:Learn how and when to remove this message
776:
561:
176:
964:
262:), the assembly programs used in these
1054:
1052:
1050:
767:. For a list of mapping aligners, see
725:file to get the best assembly contigs.
7:
735:graph and overlapping. Read length,
663:to assemble a contiguous from reads.
459:have error rates between 0.5 and 10%
141:and merging fragments from a longer
55:adding citations to reliable sources
894:http://cab.spbu.ru/software/spades/
25:
1592:www.bioinformatics.babraham.ac.uk
355:post-transcriptional modification
1262:Briefings in Functional Genomics
949:List of sequenced animal genomes
316:segments to any specific region.
31:
864:and reads with 15% error rate.
852:https://github.com/lh3/minimap2
42:needs additional citations for
939:De novo transcriptome assembly
495:de novo transcriptome assembly
363:de novo transcriptome assembly
351:single-nucleotide polymorphism
195:de novo transcriptome assembly
1:
1630:10.1093/bioinformatics/btr477
1517:10.1093/bioinformatics/btv351
1120:10.1126/science.287.5461.2196
500:Later, new technologies like
1381:10.1016/j.humimm.2021.02.012
450:are about 800–900 bases long
1227:Briefings in Bioinformatics
979:Briefings in Bioinformatics
929:De novo sequence assemblers
765:De novo sequence assemblers
369:De-novo vs mapping assembly
191:de novo sequence assemblers
1679:
912:https://samtools.github.io
741:Next Generation Sequencing
672:Types of Sequence Assembly
181:Types of sequence assembly
1312:10.5858/arpa.2012-0463-RA
1015:Baker M (27 March 2012).
446:projects where the reads
357:. Beginning in 2008 when
778:Sequence Assembly Tools
383:Overlap/Layout/Consensus
890:Short & Long reads
828:Short & Long reads
695:Bioinformatics pipeline
684:Merge chosen fragments.
620:Drosophila melanogaster
297:Drosophila melanogaster
1663:DNA sequencing methods
1457:Nucleic Acids Research
418:Technological advances
326:Expressed sequence tag
317:
182:
954:Plant genome assembly
314:
180:
1544:Nature Biotechnology
1203:amos.sourceforge.net
748:Comparative genomics
480:subsequently coined
343:alternative splicing
51:improve this article
1469:10.1093/nar/gkp1137
1274:10.1093/bfgp/elr035
1102:2000Sci...287.2196M
1096:(5461): 2196–2204.
908:Alignment analysis
779:
647:Assembly algorithms
564:
518:Nanopore sequencing
433:dideoxy termination
66:"Sequence assembly"
1420:10.1101/gr.6435207
1239:10.1093/bib/bbl015
1033:10.1038/nmeth.1935
991:10.1093/bib/bbw096
934:Sequence alignment
777:
562:
506:Applied Biosystems
444:shotgun sequencing
395:Greedy graph-based
336:housekeeping genes
318:
275:computing clusters
240:sequence alignment
183:
152:shotgun sequencing
1623:(20): 2790–2796.
1511:(19): 3210–3212.
1414:(11): 1697–1706.
1341:groups.google.com
1167:10.1101/gr.208902
944:Set cover problem
920:
919:
644:
643:
473:454 Life Sciences
439:Sanger sequencing
412:only its two ends
135:sequence assembly
127:
126:
119:
101:
16:(Redirected from
1670:
1643:
1642:
1632:
1608:
1602:
1601:
1599:
1598:
1584:
1578:
1577:
1567:
1556:10.1038/nbt.2023
1535:
1529:
1528:
1500:
1491:
1490:
1480:
1463:(6): 1767–1771.
1448:
1442:
1441:
1431:
1399:
1393:
1392:
1369:Human Immunology
1364:
1351:
1350:
1348:
1347:
1333:
1324:
1323:
1306:(9): 1296–1303.
1295:
1286:
1285:
1257:
1251:
1250:
1222:
1213:
1212:
1210:
1209:
1195:
1189:
1188:
1178:
1146:
1140:
1139:
1113:
1085:
1079:
1078:
1076:
1074:
1065:
1056:
1045:
1044:
1012:
1003:
1002:
974:
780:
763:assemblers, see
565:
467:By 2004 / 2005,
374:complexity of O(
122:
115:
111:
108:
102:
100:
59:
35:
27:
21:
1678:
1677:
1673:
1672:
1671:
1669:
1668:
1667:
1648:
1647:
1646:
1610:
1609:
1605:
1596:
1594:
1586:
1585:
1581:
1550:(11): 987–991.
1537:
1536:
1532:
1502:
1501:
1494:
1450:
1449:
1445:
1408:Genome Research
1401:
1400:
1396:
1375:(11): 801–811.
1366:
1365:
1354:
1345:
1343:
1335:
1334:
1327:
1297:
1296:
1289:
1259:
1258:
1254:
1224:
1223:
1216:
1207:
1205:
1197:
1196:
1192:
1155:Genome Research
1148:
1147:
1143:
1087:
1086:
1082:
1072:
1070:
1063:
1058:
1057:
1048:
1014:
1013:
1006:
976:
975:
966:
962:
925:
862:Oxford Nanopore
759:For a lists of
757:
697:
657:de Bruijn Graph
649:
526:
524:Quality control
489:From 2006, the
483:hybrid assembly
455:cloning vectors
420:
389:de Bruijn Graph
377:
371:
323:
302:D. melanogaster
288:DNA read errors
264:genome projects
236:
231:
175:
159:gene transcript
123:
112:
106:
103:
60:
58:
48:
36:
23:
22:
18:Genome assembly
15:
12:
11:
5:
1676:
1674:
1666:
1665:
1660:
1658:Bioinformatics
1650:
1649:
1645:
1644:
1617:Bioinformatics
1603:
1579:
1530:
1505:Bioinformatics
1492:
1443:
1394:
1352:
1325:
1287:
1252:
1214:
1190:
1161:(1): 177–189.
1141:
1111:10.1.1.79.9822
1080:
1046:
1027:(4): 333–337.
1021:Nature Methods
1004:
963:
961:
958:
957:
956:
951:
946:
941:
936:
931:
924:
921:
918:
917:
914:
909:
906:
900:
899:
896:
891:
888:
882:
881:
878:
873:
870:
866:
865:
854:
849:
846:
842:
841:
834:
829:
826:
822:
821:
806:
801:
798:
794:
793:
790:
789:Tool web page
787:
784:
756:
753:
752:
751:
744:
728:
727:
726:
719:
696:
693:
689:
688:
685:
682:
679:
668:
667:
664:
648:
645:
642:
641:
638:
635:
632:
629:
626:
623:
615:
614:
611:
608:
605:
602:
599:
596:
588:
587:
584:
581:
578:
575:
572:
569:
525:
522:
469:pyrosequencing
461:
460:
457:
451:
419:
416:
400:
399:
392:
386:
375:
370:
367:
347:trans-splicing
322:
319:
292:
291:
285:
278:
244:DNA sequencers
235:
232:
230:
227:
214:
213:
201:
198:
174:
171:
147:DNA sequencing
131:bioinformatics
125:
124:
39:
37:
30:
24:
14:
13:
10:
9:
6:
4:
3:
2:
1675:
1664:
1661:
1659:
1656:
1655:
1653:
1640:
1636:
1631:
1626:
1622:
1618:
1614:
1607:
1604:
1593:
1589:
1583:
1580:
1575:
1571:
1566:
1561:
1557:
1553:
1549:
1545:
1541:
1534:
1531:
1526:
1522:
1518:
1514:
1510:
1506:
1499:
1497:
1493:
1488:
1484:
1479:
1474:
1470:
1466:
1462:
1458:
1454:
1447:
1444:
1439:
1435:
1430:
1425:
1421:
1417:
1413:
1409:
1405:
1398:
1395:
1390:
1386:
1382:
1378:
1374:
1370:
1363:
1361:
1359:
1357:
1353:
1342:
1338:
1332:
1330:
1326:
1321:
1317:
1313:
1309:
1305:
1301:
1294:
1292:
1288:
1283:
1279:
1275:
1271:
1267:
1263:
1256:
1253:
1248:
1244:
1240:
1236:
1232:
1228:
1221:
1219:
1215:
1204:
1200:
1194:
1191:
1186:
1182:
1177:
1172:
1168:
1164:
1160:
1156:
1152:
1145:
1142:
1137:
1133:
1129:
1125:
1121:
1117:
1112:
1107:
1103:
1099:
1095:
1091:
1084:
1081:
1069:
1062:
1055:
1053:
1051:
1047:
1042:
1038:
1034:
1030:
1026:
1022:
1018:
1011:
1009:
1005:
1000:
996:
992:
988:
984:
980:
973:
971:
969:
965:
959:
955:
952:
950:
947:
945:
942:
940:
937:
935:
932:
930:
927:
926:
922:
915:
913:
910:
907:
905:
902:
901:
897:
895:
892:
889:
887:
884:
883:
879:
877:
874:
871:
868:
867:
863:
859:
855:
853:
850:
847:
844:
843:
839:
835:
833:
830:
827:
824:
823:
819:
815:
811:
807:
805:
802:
799:
796:
795:
791:
788:
785:
782:
781:
775:
772:
770:
766:
762:
754:
749:
745:
742:
738:
734:
729:
724:
720:
717:
713:
709:
705:
704:
702:
701:
700:
694:
692:
686:
683:
680:
677:
676:
675:
673:
665:
662:
658:
654:
653:
652:
646:
639:
636:
633:
630:
627:
624:
622:
621:
617:
616:
612:
609:
606:
603:
600:
597:
595:
594:
590:
589:
585:
582:
579:
576:
573:
570:
567:
566:
560:
558:
554:
550:
546:
542:
538:
537:transcriptome
533:
531:
523:
521:
519:
515:
511:
507:
503:
498:
496:
492:
487:
485:
484:
478:
474:
470:
465:
458:
456:
452:
449:
448:
447:
445:
441:
440:
435:
434:
430:In 1975, the
428:
424:
417:
415:
413:
409:
404:
396:
393:
390:
387:
384:
381:
380:
379:
368:
366:
364:
360:
356:
352:
348:
344:
339:
337:
332:
327:
320:
313:
309:
307:
303:
299:
298:
289:
286:
283:
279:
276:
272:
269:
268:
267:
265:
261:
257:
253:
249:
245:
241:
233:
228:
226:
223:
219:
211:
207:
202:
199:
196:
192:
188:
187:
186:
179:
172:
170:
166:
164:
160:
156:
153:
148:
144:
140:
136:
132:
121:
118:
110:
99:
96:
92:
89:
85:
82:
78:
75:
71:
68: –
67:
63:
62:Find sources:
56:
52:
46:
45:
40:This article
38:
34:
29:
28:
19:
1620:
1616:
1606:
1595:. Retrieved
1591:
1582:
1547:
1543:
1533:
1508:
1504:
1460:
1456:
1446:
1411:
1407:
1397:
1372:
1368:
1344:. Retrieved
1340:
1303:
1299:
1268:(1): 25–37.
1265:
1261:
1255:
1230:
1226:
1206:. Retrieved
1202:
1193:
1158:
1154:
1144:
1093:
1089:
1083:
1071:. Retrieved
1067:
1024:
1020:
985:(1): 23–40.
982:
978:
838:command line
773:
760:
758:
698:
690:
671:
669:
650:
618:
593:Homo sapiens
591:
555:and 429 for
534:
527:
499:
488:
482:
466:
462:
438:
436:method (AKA
432:
429:
425:
421:
405:
401:
394:
388:
382:
372:
340:
324:
301:
295:
293:
281:
258:and finally
237:
215:
184:
167:
134:
128:
113:
107:October 2017
104:
94:
87:
80:
73:
61:
49:Please help
44:verification
41:
1233:(1): 6–21.
1199:"AMOS WIKI"
872:Long reads
848:Long reads
551:, 1438 for
543:, 2675 for
541:vertebrates
510:Ion Torrent
308:framework.
306:open source
222:short reads
1652:Categories
1597:2022-05-09
1346:2023-01-02
1208:2023-01-02
960:References
836:This is a
786:Read type
557:eukaryotes
547:, 843 for
545:arthropods
260:eukaryotes
229:Assemblies
218:long reads
210:long-reads
137:refers to
77:newspapers
1106:CiteSeerX
1041:1548-7105
845:MiniMap2
800:Multiple
783:Software
733:de Bruijn
708:base call
549:metazoans
271:terabytes
220:to mimic
1639:21856737
1574:22068540
1525:26059717
1487:20015970
1438:17908823
1389:33745759
1320:23991743
1282:22184334
1247:16772268
1185:11779843
1128:10731133
1059:Wolf B.
999:27742661
923:See also
904:Samtools
869:LoReTTA
810:Illumina
755:Programs
737:coverage
568:Species
491:Illumina
398:overlap.
256:bacteria
252:plasmids
157:DNA, or
139:aligning
1565:5531759
1478:2847217
1429:2045152
1136:6049420
1098:Bibcode
1090:Science
1073:6 April
797:FastQC
761:de-novo
625:13,918
598:20,364
477:Newbler
359:RNA-Seq
282:repeats
248:viruses
155:genomic
91:scholar
1637:
1572:
1562:
1523:
1485:
1475:
1436:
1426:
1387:
1318:
1280:
1245:
1183:
1176:155255
1173:
1134:
1126:
1108:
1039:
997:
886:SPAdes
860:&
858:PacBio
818:PacBio
816:, and
792:Notes
716:PacBio
661:k-mers
640:2,675
613:3,023
571:genes
353:, and
234:Genome
93:
86:
79:
72:
64:
1132:S2CID
1064:(PDF)
723:FASTQ
712:Phred
553:fungi
530:Phred
504:from
502:SOLiD
408:graph
250:over
206:k-mer
173:Types
98:JSTOR
84:books
1635:PMID
1570:PMID
1521:PMID
1483:PMID
1434:PMID
1385:PMID
1316:PMID
1278:PMID
1243:PMID
1181:PMID
1124:PMID
1075:2019
1037:ISSN
995:PMID
825:BWA
637:0.0
634:0.2
631:3.7
610:0.0
607:0.0
604:1.7
514:SMRT
512:and
331:mRNA
163:ESTs
70:news
1625:doi
1560:PMC
1552:doi
1513:doi
1473:PMC
1465:doi
1424:PMC
1416:doi
1377:doi
1308:doi
1304:137
1270:doi
1235:doi
1171:PMC
1163:doi
1116:doi
1094:287
1029:doi
987:doi
814:454
674:):
628:99
601:99
586:n:
583:M:
580:F:
577:D:
574:C:
321:EST
254:to
165:).
143:DNA
129:In
53:by
1654::
1633:.
1621:27
1619:.
1615:.
1590:.
1568:.
1558:.
1548:29
1546:.
1542:.
1519:.
1509:31
1507:.
1495:^
1481:.
1471:.
1461:38
1459:.
1455:.
1432:.
1422:.
1412:17
1410:.
1406:.
1383:.
1373:82
1355:^
1339:.
1328:^
1314:.
1302:.
1290:^
1276:.
1266:11
1264:.
1241:.
1229:.
1217:^
1201:.
1179:.
1169:.
1159:12
1157:.
1153:.
1130:.
1122:.
1114:.
1104:.
1092:.
1066:.
1049:^
1035:.
1023:.
1019:.
1007:^
993:.
983:19
981:.
967:^
820:.
812:,
771:.
508:,
486:.
365:.
349:,
345:,
193:,
133:,
1641:.
1627::
1600:.
1576:.
1554::
1527:.
1515::
1489:.
1467::
1440:.
1418::
1391:.
1379::
1349:.
1322:.
1310::
1284:.
1272::
1249:.
1237::
1231:8
1211:.
1187:.
1165::
1138:.
1118::
1100::
1077:.
1043:.
1031::
1025:9
1001:.
989::
376:n
277:;
212:.
197:)
161:(
120:)
114:(
109:)
105:(
95:·
88:·
81:·
74:·
47:.
20:)
Text is available under the Creative Commons Attribution-ShareAlike License. Additional terms may apply.